
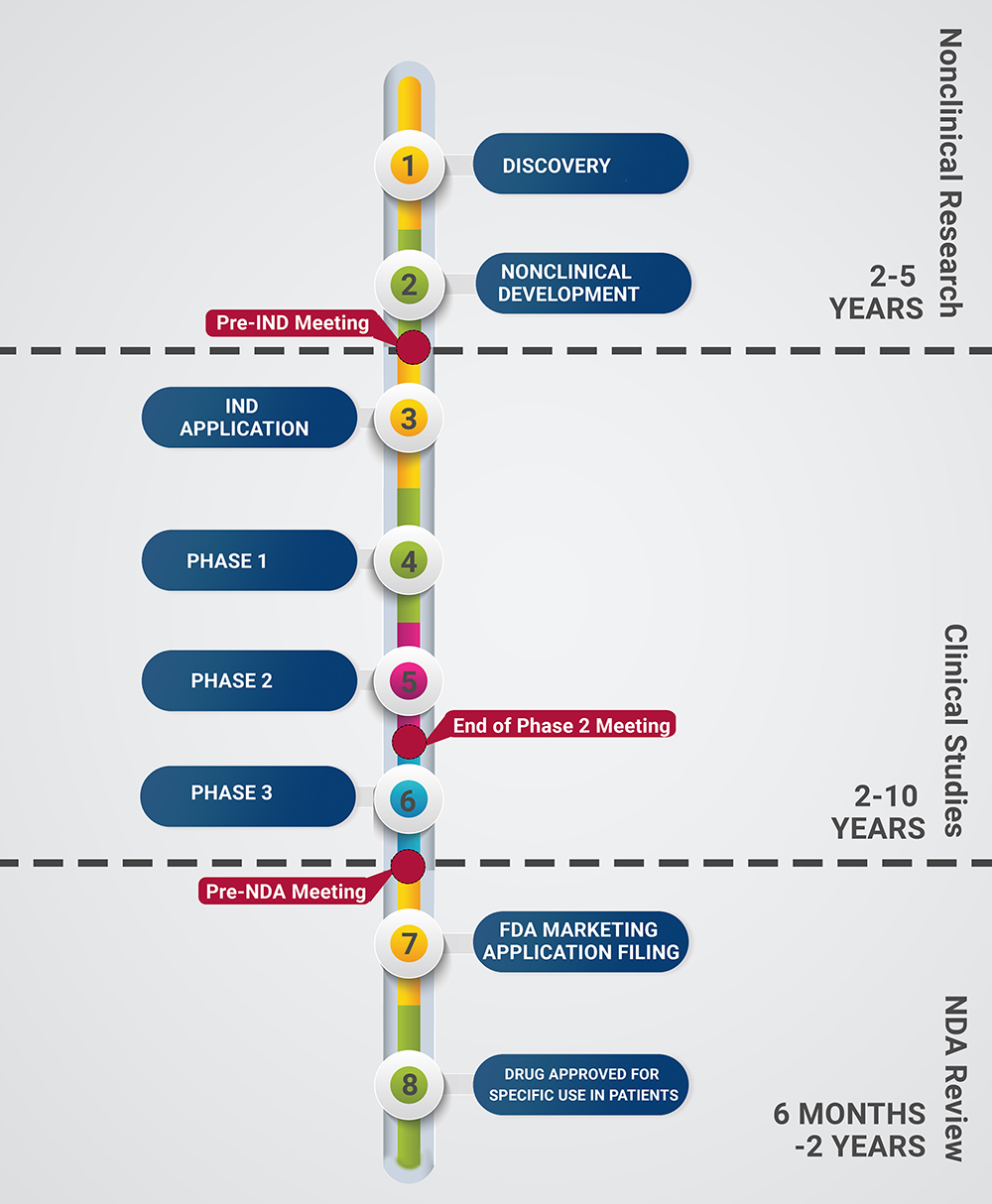
Bringing an investigational drug from bench to bedside is a long, complicated process involving many nonclinical and clinical investigations and layers of regulatory oversight. This page provides an easy-to-follow timeline for the life cycle of a new drug and detailed descriptions for the process.
Discovery
Targets can be either proteins, genes, or RNAs, and must be:
- accessible to the putative drug molecule
- elicit a biological response upon binding, and
- the biological response must be measurable in vitro and in vivo
- Data mining (bioinformatics approach)
- Publications
- Patents
- Gene Expression Data
- Proteomics Data
- Transgenic Phenotyping
- Compound profiling data
- Examination of mRNA/Protein Levels to Determine Expression in a Disease and Correlation with Disease Exacerbation or Progression
- Genetic Associations-(links between genetic polymorphism and risk of disease or disease progression)
- Phenotypic Screening (e.g. phage display)
- Small bioactive molecules that interact with and functionally modulated effector proteins
- Antisense technology
- Transgenic animals
- gene knock-outs
- gene knock-ins (non-enzymatically functioning protein replaces endogenous protein
- siRNA
- Monoclonal antibodies for cell surface and secreted proteins
- Biological Assay Development to Identify Molecules with Activity at Target
- Screening of Compound Libraries (1 in 10,000 makes it to market)
- High Throughput
- Focused Screen
- Fragment Screen
- Structural Aided Drug Design
- Virtual Screen
- Physiological Screen
- NMR Screen
- Defining a Hit Series
- Refine "hit series" of compounds to produce more potent and selective compounds with PK properties adequate to test in in vivo models
- Intensive Structure Activity Relationship (SAR) investigations
Nonclinical Development
- Understanding the clinical trial design is critical in the design of the nonclinical studies.
- Minimally, knowledge of the clinical dosing route, frequency, duration, and anticipated dose range are needed to adequately design the nonclinical studies.
- Overall purpose of nonclinical development is to provide information regarding:
- Exposure response relationship (a drug can be determined to be safe and effective only when the relationship of beneficial and adverse effects to a defined exposure is known).
- Potential drug toxicities
- Safety margins
- Estimates Identification of target organ toxicities
- Evaluation of reversibility of observed effects
- Issues to consider
- Species difference in absorption, distribution, metabolism and excretion (ADME) of the specific drug (see below for further information about ADME)
- Expression of relevant receptors or epitopes in a specific species
- Pharmacological activity
- Bioavailability
- Small Molecules-'Most Appropriate Species'
- in vitro metabolic profiling of products produced from the drug (liver microsomes from a full spectrum of species-mouse, rat, dog, mini-pig, nonhuman primate, human)
- Biopharmaceuticals-'Most Relevant Species'
- in vitro and in vivo assays that demonstrate the ability of the drug to elicit a pharmacological effect
- Difference in species selection for toxicology studies versus nonclinical development (research of your drug). See below.
It is important to develop and qualify/validate these assays early in the development process for concentration and stability, homogeneity as necessary.
- Methods required to determine the amount of drug in the preparations administered to animals
- One method for each vehicle used-i.e. one method for general toxicology and safety pharmacology studies, one method for in vitro genetic toxicology studies, one method for hERG assay.
- Methods required to determine the amount of drug in serum/plasma in animals
- One method for rodent, nonrodent and human
- Methods required to determine the presence of antibodies against the drug in serum of animals for biopharmaceuticals
- Primary PD studies (in vivo and/or in vitro) are intended to investigate the mode of action and/or effects of a substance in relation to its desired therapeutic target.
- Such studies are generally conducted during the discovery phase of pharmaceutical development and as such, are not generally conducted in accordance with Good Laboratory Practices (GLP).
- DMPK is how drug is absorbed, where it enters the body, what it breaks down into and how the body eliminates it.
- Investigations to provide information on
- Systemic exposure to the animals by measurement of blood levels (=Absorption)
- Distribution and retention within major organs of the rodent by whole body autoradiography (=Distribution)
- Major metabolic pathways through in vitro and in vivo investigations for demonstration of major metabolic similarity / dissimilarity between the animal species used in toxicity studies and humans (=Metabolism)
- Rate and routes of elimination (=Excretion)
- Absorption
- Single dose Pharmacokinetics (p.o.; i.v.) in several animal species
- Distribution
- Mass balance
- Quantitative whole-body autoradiography (QWBA)
- Protein binding
- Red cell partitioning and brain penetration
- Metabolism (stability & pattern; identification)
- in vitro: microsomes; hepatocytes of several species
- CYP induction (metabolizing enzymes and transporters)
- in vivo metabolism
- Excretion
- Biliary; Urinary
- Screen of different Formulation
- Bioavailibility
After primary pharmacology and basic ADME data in animals are available but before performance of toxicology studies to support an IND, it is time to review with the FDA during a pre-IND meeting the overall plan for the toxicology studies, the initial clinical plan and the basic manufacturing process(es). More information can be found on our pre-IND page.
- The design of any nonclinical package to support an IND or BLA submission depends on test article type, indication, route, and the design of the initial clinical plan.
- These studies must be performed in compliance with Good Laboratory Practices (GLPs).
- Dose Selection-a control and three dose levels are generally evaluated.
- Low dose should provide No Observed Adverse Effect Level (NOAEL) and generally selected based on an acceptable margin of safety with regard to the proposed clinical starting dose.
- The high dose should define:
- The Maximum Tolerated Dose (MTD)-the highest dose of a drug or treatment that does not cause unacceptable side effects and identify target organ toxicities; or
- The Maximum Feasible Dose (MFD)- limiting dose that achieves large exposure multiples or saturation of exposure
- The mid dose should characterize the dose-response relationship
- Non-GLP Dose Range Finding (DRF) study in rodents and nonrodents
- Limited number of animals
- Route of administration the same as proposed for clinical studies
- Dose level escalated to define MTD in single dose and short repeat dose phase
- GLP Repeat Dose Toxicology Studies
- Larger number of animals to provide statistical power
- Route of administration the same as proposed for clinical studies
- Dose levels dependent on data from DRF studies
- Duration dependent on design of Clinical Trial
- Toxicokinetics
- Recovery arm to determine whether effects increase, decrease or remain the same following cessation of dosing
- Other studies dependent on the route of administration
- Local tolerance studies
- Delayed hypersensitivity studies (dermal products)
- Hemocompatiblity (intravenous products)
- Carcinogenicity
- Assessment of drug to exert a pharmacological effect on critical organ systems, generally at lower dose than toxicology studies to identify subtle effects.
- These studies must be performed in compliance with Good Laboratory Practices (GLPs)
- Cardiovascular in nonrodent
- Same species as nonrodent species in toxicology studies
- Telemetry of animals
- Collection of data for 24 hours following dose administration
- in vitro hERG
- Assay determines possible interaction of drug with the hERG potassium channel on mammalian hearts
- Blockage of hERG channel can cause QT prolongation which can lead to ventricular arrhythmia (Torsades de Pointes)
- Central Nervous System Study in Rodents
- Same rodent specifies as selected for the toxicology studies
- Evaluation of locomotion, grip strength, hind-limb splay, pain perception, reaction to stimuli etc.
- Respiratory Study in Rodents
- Same rodent specifies as selected for the toxicology studies
- Rodents placed in plethysmograph (instrument for measuring changes in volume within an organ or whole body usually resulting from fluctuations in the amount of blood or air it contains).
- Measure respiratory rate, tidal volume, minute volume etc.
- Other studies depending on pharmacology of the drug may be required
- Renal Safety Pharmacology
- Gastrointestinal Motility
- GLP compliant
- Ames test-bacterial assay to determine if drug can cause point mutations
- Chromosomal aberration
- in vitro assay with S9 metabolic system
- Evaluates the potential for the drug to damage chromosomes (clastogenicity)
- in vivo Micronucleus test in rats/ mice
- in vivo study to determine drugs potential for clastogenicity
- Can be deferred to Phase 2 but generally done along with other studies