
After the IDE submission has been delivered to the FDA, it undergoes a review process with several possible outcomes.
Once the initial IDE submission has been sent to the FDA, a team of staff reviews the IDE and provides one of several standard responses within 30 days of receipt. For additional information, see FDA's Guidance document entitled FDA Decisions for Investigational Device Exemption Clinical Investigations.
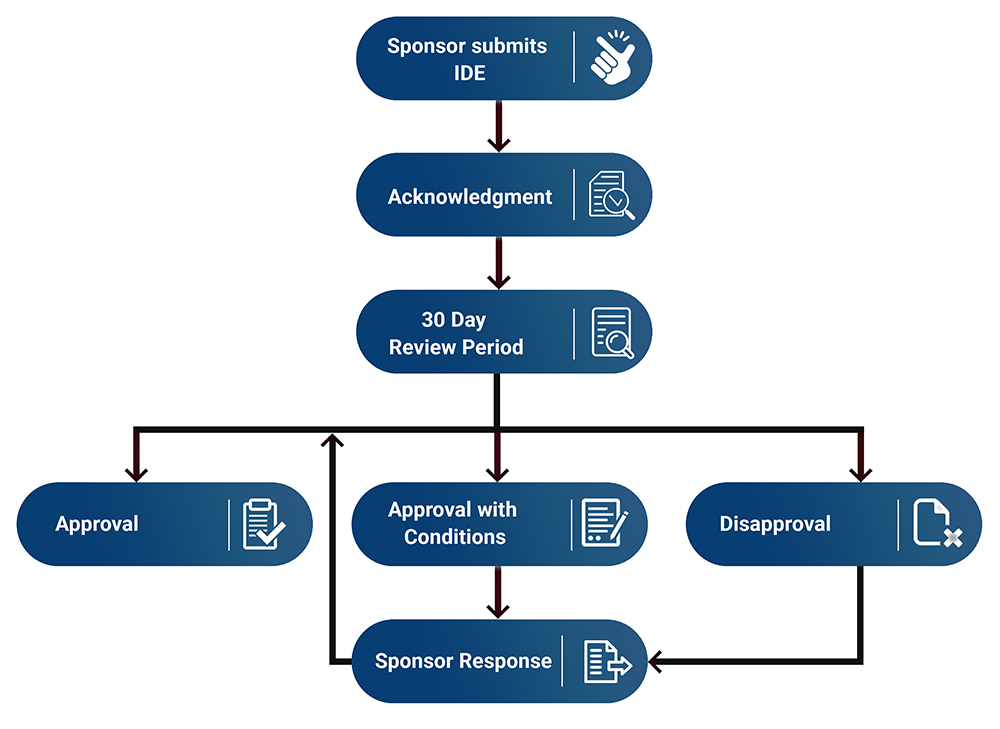
Approval: If FDA approves an IDE application, the sponsor may begin subject enrollment upon receipt of IRB approval and in accordance with the limits described in FDA's decision letter, including the maximum numbers of U.S. subjects and investigational sites.
Note: If you have not heard back from the FDA within 30 days, it is recommended that the submitter send an email to the contact named on the FDA acknowledgement confirming the study can proceed.
An IDE application is approved if FDA has determined that: the sponsor has provided sufficient data to support initiation of a human clinical study; no subject protection concerns preclude initiation of the investigation; and no additional conditions must be met.
In some cases, FDA may determine that an outstanding issue remains that can be addressed with data that will be gathered concurrently with the enrollment of a portion of study subjects (i.e., staged approval). FDA may also inform the sponsor of recommended modifications to the study design that FDA believes will improve the study and may be necessary to enable the study to support a future marketing application (i.e., study design considerations) as well as other issues that FDA believes should be considered in preparing for a marketing application or a future clinical investigation (i.e., future considerations).
Approval with Conditions: If FDA approves an IDE application with conditions, the sponsor may begin subject enrollment upon receipt of IRB approval with start date, but there may be limits described in FDA's decision letter, including the maximum numbers of U.S. subjects and investigational sites. If applicable, within 45 days the Sponsor must submit information via an amendment addressing the issues identified as conditions of approval in FDA's decision letter. Outstanding issues that may lead to approval with conditions include, but are not limited to:
- Requests for additional information or data involving non-clinical testing issues that do not need to be resolved prior to initiation of subject enrollment;
- Late-stage follow-up procedures and assessments that relate to the care of study subjects but, because they occur late in the study, can likely be addressed in response to FDA's letter prior to subjects' reaching that point in the study;
- Minor issues related to the informed consent document that must be corrected before initiation of subject enrollment but FDA can review after implementation;
- Other minor clarifications, corrections, or modifications (not related to study design) that do not need to be resolved prior to initiation of subject enrollment.
The FDA will inform the Sponsor of its decision 30 days after receipt of the amendment. During this time, the Sponsor may continue to conduct the study within the limitations defined in FDA's decision letter. If FDA determines that the issues have been adequately resolved, it will grant approval.
If any issues remain, FDA may again grant approval with conditions and will communicate those remaining outstanding issues to the Sponsor by letter. In this case, the Sponsor may continue to enroll subjects in the study provided that, within another 45 days, the sponsor responds to the remaining issues identified in FDA's letter. If the sponsor's response to FDA's questions raises concerns regarding subject safety, or the sponsor does not respond, FDA may take appropriate regulatory actions to protect study subjects, including placing a clinical hold on the study. If the study is placed on hold, no additional subjects may be enrolled, and previously enrolled study subjects should receive appropriate monitoring and treatment for their safety.
Disapproval: If an IDE application is disapproved, the sponsor may not initiate enrollment in the clinical investigation until the sponsor submits an amendment to the IDE to respond to the deficiencies identified in FDA's letter and subsequently receives a new letter from FDA granting approval or approval with conditions.
Consistent with 21 CFR 812.30(b) and section 520(g) of the FD&C Act, FDA may disapprove an IDE for any of the following reasons:
- Failure to comply with any requirement in 21 CFR Part 812 or section 520(g) of the FD&C Act, any other applicable regulation or statute, or any condition of approval imposed by an IRB or FDA. (21 CFR 812.30(b)(1)).
- The application or a report contains an untrue statement of material fact, or omits material information required by 21 CFR Part 812. (21 CFR 812.30(b)(2)).
- The sponsor fails to respond to a request for additional information within the time FDA prescribes. (21 CFR 812.30(b)(3)).
- There is reason to believe that risks to the subjects are not outweighed by the anticipated benefits to the subjects and the importance of the knowledge to be gained (21 CFR 812.30(b)(4)).
- The informed consent is inadequate. (21 CFR 812.30(b)(4)).
- The investigation, as proposed, is scientifically unsound. (21 CFR 812.30(b)(4)).
- There is reason to believe that the device as used is ineffective. (21 CFR 812.30(b)(4)).
- It is otherwise unreasonable to begin or to continue the investigation owing to the way in which the device is used or the inadequacy of (i) the report of prior investigations or the investigational plan; (ii) the methods, facilities, and controls used for the manufacturing, processing, packaging, storage, and where appropriate, installation of the device; or (iii) monitoring and review of the investigation. (21 CFR 812.30(b)(5)).
The Q-Submission Program Overview: Requests for Feedback and Meetings for Medical Device Submissions
The FDA encourages sponsors of clinical trials testing medical devices to communicate openly with the Agency. In relation to IDEs, the Q-Submission Program, which replaced the pre-IDE program in 2017, is used to provide mechanisms for investigators and sponsors to request feedback from, or a meeting with, the FDA regarding potential or planned medical devices. The program supports the following types of medical device submissions:
- Investigational Device Exemption (IDE) Applications,
- Premarket Approval (PMA) Applications,
- Humanitarian Device Exemptions (HDE) Applications,
- Evaluation of Automatic Class III Designations (De Novo requests),
- Premarket Notification (510(k)) Submissions,
- Clinical Laboratory Improvement Amendments (CLIA) Waiver by Applications (CW),
- Dual 510(k) and CLIA Waiver by Application Submissions (Duals),
- Accessory Classification Requests, and
- Certain Investigational New Drug Applications (INDs) and Biologics License Applications (BLAs) submitted to the Center for Biologics Evaluation and Research (CBER).
By communicating with FDA throughout the submission process, information can be exchanged in a more expeditious manner thus reducing the review cycle turn-around time. FDA provides advice to sponsors through the Q-Submission program. A Q-Sub request must include one electronic copy (eCopy) in the English language. For information regarding eCopy, refer to the Initial IDE submission section. For more information about the Q-submission process or any of the specific Q-Subs described below, including content requirements, please see FDA's Guidance entitled "Requests for Feedback on Medical Device Submissions: The Pre-Submission Program and Meetings with Food and Drug Administration Staff" and the subsequent draft June 7, 2018, guidance "Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program".
- Informational Meeting: Informational meetings are appropriate when a sponsor wants to share information with FDA without an expectation of feedback. FDA will be in a listening mode. This meeting type is appropriate when a sponsor wants to provide an overview of ongoing device development when there are multiple submissions planned within the year. This is also useful to familiarize the FDA review team with a new device that has significant technological differences from current devices and to educate the reviewers on advancements they will be seeing in the future.
- Agreement Meeting: Agreement meetings are available to any sponsor planning to investigate the safety or effectiveness of a class III product or any implant. The Agreement Meeting is available to submitters of 510(k)s for eligible devices as well. The purpose of this meeting is to reach agreement on the key parameters of the investigational plan (see 21 CFR 812.25), including the clinical protocol. The meeting is to be held within 30 days of FDA receipt of the request for the meeting. Any agreement reached in this meeting is also to be written, shared with the applicant, and made part of the administrative record. It is binding on the FDA and may be changed only with the written agreement of the applicant or when there is a substantial scientific issue essential to determining the safety or effectiveness of the device.
- Determination Meeting: (Please note, this is different than a Study Risk Determination or a determination by the IRB) A Determination Meeting is available to anyone anticipating submitting a PMA or Product Development Protocol (PDP) and is intended to provide the applicant with the Agency's determination of the type of valid scientific evidence that will be necessary to demonstrate that the device is effective for its intended use. This type of meeting will result in FDA's conclusion whether clinical studies are needed to establish effectiveness and, in consultation with the applicant, determine the least burdensome way of evaluating device effectiveness that has a reasonable likelihood of success. The applicant can expect that FDA will determine if concurrent randomized or non-randomized controls, historical controls, or other types of evidence will be acceptable. FDA's determination is to be written, shared with the applicant within 30 days following the meeting, and is binding upon the Agency, unless it would be contrary to public health.
- It is important to note the Agreement and Determination Meetings are intended for those studies for which the endpoint is pre-determined to be commercial product marketing approval from FDA. Additionally, both type meetings require a formal letter and meeting to be effectively utilized. For additional information regarding Agreement and Determination Meetings, please see FDA's Guidance on Early Collaboration Meetings Under the FDA Modernization Act (FDAMA).
- Day 100 Meeting: A PMA applicant may request a Day 100 Meeting to discuss the review status of their PMA. FDA will inform the applicant of any identified deficiencies prior to the meeting, which will take place no later than 100 days after the receipt of the PMA application. FDA recommends that a request for a Day 100 Meeting be submitted with the original PMA or as a Q-Submission (Q-Sub Day 100 Meeting Request) no later than 70 days from the PMA filing date so that FDA has sufficient time to schedule the meeting. This meeting may be used to discuss identified issues and remedial actions, an action plan with estimated dates of completion, FDA estimated timetables for review completion, the need for panel involvement, or possible premarket versus post market requirements.
- Pre-Submissions: Defined as a formal written request for Agency feedback, the Pre-Submission or Pre-Sub is not a required submission nor is the response received binding on the part of the FDA. A sponsor is encouraged to request a Pre-Sub when FDA's feedback on specific questions is necessary to guide product development and/or application preparation. A Pre-Sub may be submitted in advance of an IDE or marketing application, and importantly, a Pre-Sub may also be submitted for a clinical study for which an IDE would not be required, (e.g., NSR device studies, IDE exempt device studies, or for studies conducted entirely outside the USA (OUS)). The Pre-Sub should include a request for an in-person meeting, teleconference, or written responses to specific questions regarding the IDE submission and/or study protocol associated with the device. For all Pre-Submissions in which a meeting or teleconference is being requested, a minimum of three proposed meeting dates should be provided in the initial submission. Within 15 calendar days, FDA will either confirm one of the dates or provide two alternative dates prior to calendar day 75 from the receipt of the accepted submission. FDA will provide written feedback with responses to the sponsor's questions for all Pre-Subs, regardless of whether the sponsor requested a meeting, teleconference, or written feedback.
- Study Risk Determination: FDA will review a study protocol and issue a letter to the sponsor indicating if the study is exempt, or if not exempt, whether the study is significant risk or non-significant risk. This letter may be submitted to the Institutional Review Board which does not need to conduct its own independent assessment of the study risk because FDA's determination is final. Please note that a study risk determination does not obligate the sponsor to submit a future IDE application. For additional information regarding Study Risk Determinations, please refer to the FDA Information Sheet Guidance For IRBs, Clinical Investigators, and Sponsors – Significant Risk and Nonsignificant Risk Medical Device Studies.
- Submission Issue Request (SIR): A sponsor may request this type meeting to discuss deficiencies identified during FDA's review of an application. The purpose of the meeting is to request FDA feedback on a proposed approach to address issues conveyed in a marketing submission hold letter, CLIA Waiver (CW) hold letter, or an IDE Letter. The SIR is intended to facilitate interaction between FDA and the sponsor to quickly resolve or clarify issues identified that are associated with a request for additional information regarding a 510(k), De Novo request, and CWs, major deficiencies resulting in not approvable, approvable with deficiencies, approvable pending GMP, or Approval with Post-Approval Study (PAS) conditions for pre-marketing applications (PMAs) and Humanitarian Device Exemptions (HDEs). This may be addressed in a submission issue meeting or teleconference. This is not necessary for simple requests for clarification of issues in a letter that does not require the involvement of management or to discuss issues while a file is under active review. This process is also not appropriate for discussing letters conveying FDA's final decisions, such as Not Substantially Equivalent, Withdrawals, or Deletions. If a Submission Issue Request is received within 30 days of FDA's marketing submission hold, IND Clinical Hold letter, or IDE letter, the FDA team will aim to provide feedback within 21 days, as resources permit. This is referred to as a Submission Issue Request A. If a Submission Issue Request is submitted more than 30 days after FDA's letter, FDA will aim to provide feedback within 70 days, as resources permit. This is referred to as a Submission Issue Request B.
| Q-Sub Type | Method of Feedback | Timeline for Feedback (From Receipt of Submission) |
|---|---|---|
| Pre-Submission | Written Feedback Only | 70 days |
| Pre-Submission | Meeting (face-to-face or teleconference) with written feedback provided in advance | Written Feedback: 70 days or 5 days prior to scheduled meeting whichever is sooner Meeting: Date based on mutual agreement (typically at 60-75 days) |
| Submission Issue Request (SIR) | Meeting or Written Feedback | If SIR is received within 60 days of FDA's marketing submission letter: 21 days as resources permit If SIR is received more than 60 days after FDA's marketing submission letter: 70 days as resources permit |
| Study Risk Determination | Formal Letter | 90 days |
| Informational Meeting* | Meeting | 90 days |
*When used to track requests that do not meet the definition of a Q-Sub type, Informational Meeting timeframe and feedback mechanism can vary. Typically, informational meetings do not include FDA feedback.