
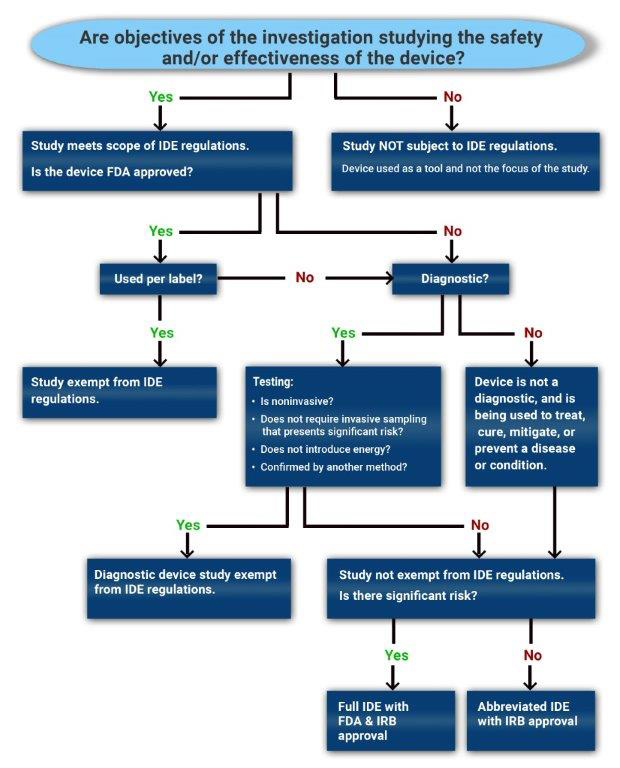
Not all clinical device studies need to operate under an IDE. Use the decision tree to determine whether a proposed investigation will require IDE submission and FDA oversight.
IDE Exemption Criteria
- If the objective of the clinical investigation is to assess the safety and/or effectiveness of a medical device, then the study is a device study and is subject to regulatory oversight by the US Food and Drug Administration as defined in 21 CFR 812 (Investigational Device Exemption).
- If the objective of the study is not to test the safety or effectiveness of the device, then the study would not fall within the scope of 21 CFR 812.
- Such a device is used as a "tool". One example of this would be when an MRI is used to collect data in an oncology drug trial to evaluate tumor response or a thermometer used to check temperature as an inclusion criterion for a study.
A device investigation is exempted from the IDE regulations if the device fits any of the following criteria (21 CFR 812.2(c)):
- A legally marketed device when used in accordance with its labeling.
- This criterion applies to devices with an approved PMA or 510(k) clearance, but does not apply to transitional devices, which are devices that were regulated by FDA as new drugs before May 28, 1976.
- A diagnostic device if it complies with the labeling requirements in §809.10(c) and if the testing (see below for more details):
- is noninvasive;
- does not require an invasive sampling procedure that presents significant risk;
- does not by design or intention introduce energy into a subject; and
- is not used as a diagnostic procedure without confirmation by another medically established diagnostic product or procedure;
- A device undergoing consumer preference testing, testing of a modification, or testing of a combination of two or more devices in commercial distribution, if the testing is not for the purpose of determining safety or effectiveness and does not put subjects at risk.
- A device intended solely for veterinary use.
- A device shipped solely for research on or with laboratory animals and labeled in accordance with 21 CFR 812.5(c).
- A custom device, as defined in 21 CFR 812.3(b), unless the device is being used to determine safety or effectiveness for commercial distribution.
- Custom devices are "limited to no more than five units per year of a particular device type" and require annual reporting to the FDA. For additional information, refer to the FDA Guidance for Industry and FDA Staff – Custom Device Exemption.
A diagnostic device is exempt from the IDE regulations, if the following four criteria accurately describe the use of the device in the planned clinical study. The device:
- Is noninvasive: "A noninvasive device is one that does not, by design or intention:
- penetrate or pierce the skin or mucous membranes of the body, the ocular cavity, or the urethra; or
- enter the ear beyond the external auditory canal, the nose beyond the nares, the mouth beyond the pharynx, the anal canal beyond the rectum, or the vagina beyond the cervical os (21 CFR 812.3(k)).”
- Does not require an invasive sampling procedure that presents significant risk: "While the device itself may be noninvasive, the sampling required to use a diagnostic device may be invasive, such as obtaining a biopsy. To determine if an invasive sampling procedure presents a significant risk:
- FDA recommends…that you base your risk determination on the nature of the harm that may result from sampling. For example, FDA considers sampling techniques that require biopsy of a major organ, use of general anesthesia, or placement of a blood access line into an artery or large vein (subclavian, femoral, or iliac) to present a significant risk.
- Blood sampling that involves simple venipuncture is considered noninvasive, and the use of surplus samples of body fluids or tissues that are left over from samples taken for non-investigational purposes is … considered noninvasive (21 CFR 812.3(k)).”
- Does not by design or intention introduce energy into a subject: "Any form of energy qualifies (e.g. light, heat, X-ray, gamma ray, magnetic fields). The study can be exempt if energy is introduced as a part of clinical care and there is no additional energy introduced because of the study. For example, if the investigational diagnostic device is applied to X-ray images collected as a part of clinical care, then the study can be exempt.”
- Is not used as a diagnostic procedure without confirmation of the diagnosis by another, medically established diagnostic product or procedure:
- Test results should not influence patient treatment or clinical management decisions before the diagnosis is established by a medically established product or procedure.
- If an investigational test uses a new technology or represents a significant technological advance, established diagnostic products or procedures may not be adequate to confirm the diagnosis provided by the investigational IVD.
- If the diagnostic is not being used to make treatment decisions for subjects in the study, then confirmation of the diagnosis is not required as a part of that study in order to meet this criterion.”
For additional information about IVDs and the diagnostic device exemption criteria, please see the FDA Guidance for Industry and FDA Staff - In Vitro Diagnostic (IVD) Device Studies - Frequently Asked Questions.
The study requires an IDE (full or abbreviated). However, in order to decide which type of IDE is needed, an SR/NSR determination is required.
- A non-significant risk (NSR) study requires an Abbreviated IDE and is solely overseen by an IRB.
- A significant risk (SR) study requires an IDE that is reviewed by the FDA as well as the IRB.