
Not all clinical studies need to operate under an IND. Use the decision tree to determine whether a proposed investigation will require IND submission and FDA oversight.
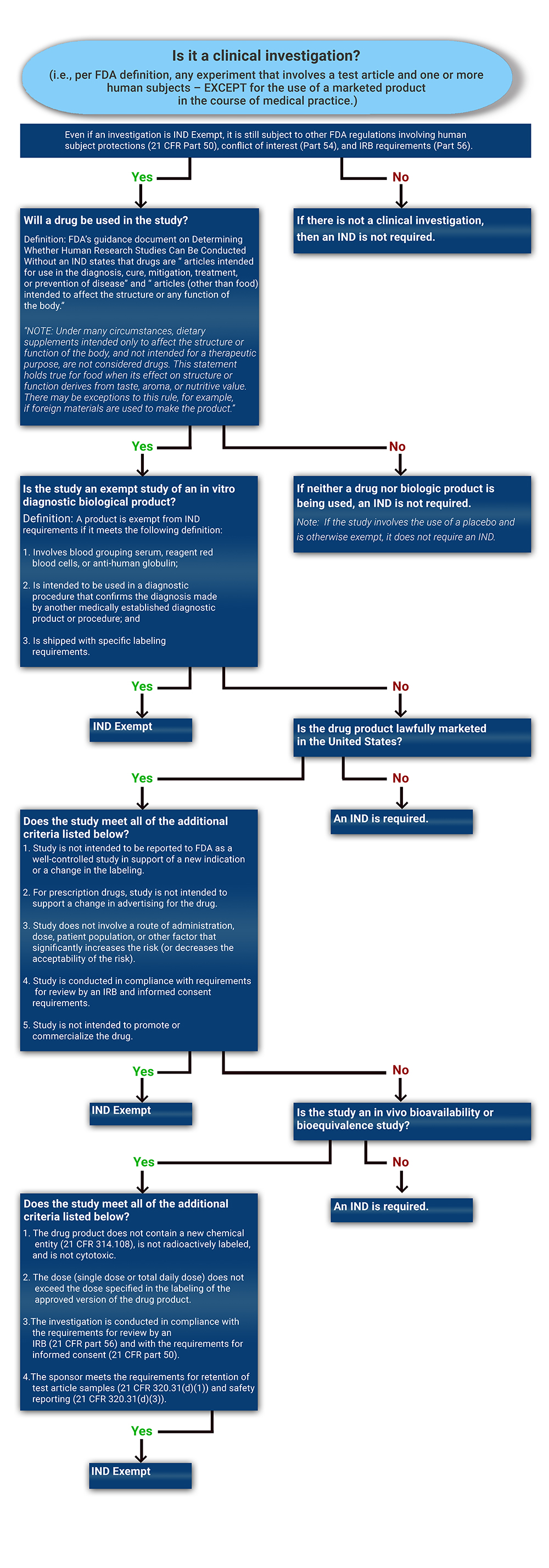
To determine if an IND is required for a clinical study, please start by reviewing the decision tree. The following FDA Guidance document is an excellent source of information that should also be carefully reviewed: Guidance for Clinical Investigators, Sponsors, and IRBs Investigational New Drug Applications (INDs) — Determining Whether Human Research Studies Can Be Conducted Without an IND, September 2013".
In general, the IND regulations in part 312 require that human research studies be conducted under an IND if all of the following conditions exist:
- The research involves a drug as that term is defined in section 201(g)(1) of the FD&C Act (21 U.S.C. 321(g)(1)).
- The research is a clinical investigation as defined in the IND regulations (21 CFR 312.3).
- The clinical investigation is not otherwise exempt from the IND requirements in part 312. There are a few exceptions when an IND may not be needed for a clinical study when utilizing an FDA-regulated product. Each NIH Institute has a regulatory group or professional who can help assess whether an IND is needed for your study. Please refer to our IC Regulatory Contacts page for our assistance at any point during the process. Although not required, Sponsors looking for FDA guidance to confirm the requirements for the development process may request a pre-IND meeting with the FDA.
Whether a clinical investigation of a marketed drug can be eligible for an IND exemption primarily depends on the intent of the investigation and the degree of risk associated with the use of the drug in the investigation. If the drug is not legally marketed, the study is not eligible for IND exemption and would require an IND. For additional information please review The FDA Guidance: Investigational New Drug Applications (INDs) — Determining Whether Human Research Studies Can Be Conducted Without an IND.
A clinical investigation using a drug product that is lawfully marketed in the United States is exempt from the IND requirements if all of the criteria for an exemption in §312.2(b) are met:
- The investigation does not involve a route of administration, dose, patient population, or other factor that significantly increases the risk (or decreases the acceptability of the risk) associated with the use of the drug product (21 CFR 312.2(b)(1)(iii)).
- The investigation is not intended to be reported to FDA as a well-controlled study in support of a new indication and there is no intent to use it to support any other significant change in the labeling of the drug.
- The investigation is conducted in compliance with the requirements of §312.7 (i.e., the investigation is not intended to promote or commercialize the drug product).
- In the case of a prescription drug, the investigation is not intended to support a significant change in the advertising for the drug.
- The investigation is conducted in compliance with the requirements for review by an IRB (21 CFR part 56) and with the requirements for informed consent (21 CFR part 50).
What Is Meant by a Drug Product That Is Lawfully Marketed in the United States?
A product must be legally marketed as a drug, meaning it has an NDA, an Abbreviated New Drug Application (ANDA), or a Biologics Licensing Application (BLA) that has previously been approved by the FDA, in order to be considered lawfully marketed in the United State. The labeling or package insert details the approved indications for use and dosage and administration. The most up-to-date drug labeling information can be found via the DailyMed database.
A study may be exempt from IND requirements even if the drug is not used in exactly the same dosage form, dosage levels, and patient populations described in the marketed labeling for the product. Changes to the lawfully marketed drug product that do not increase the risks over the risk presented by use of the product are allowed. Sponsors can make low-risk modifications to the lawfully marketed dosage form to, for example, blind a study using over-encapsulation of a tablet. Questions regarding modifications should be directed to your IC’s regulatory professional.
The Sponsor must also determine their intent to report an investigation as a well-controlled study to the FDA. Will the sponsor be submitting to FDA
- in support of a new indication?
- in support of any other significant change in the labeling of the drug? or
- in support of a significant change in the advertising for the drug (for prescription drugs only)?
Generally, any well-controlled trial of a marketed drug (e.g., a study of a new indication) sponsored by the manufacturer of the drug would be intended to be used to influence labeling or promotion in some significant way and would have to be conducted under an IND.
Studies of marketed drugs conducted by an entity that does not have an independent ability to change a drug's labeling – e.g., a study initiated by a Principal Investigator and sponsored by an IC at the NIH – would not generally be intended to be submitted to FDA to support a new indication or to otherwise influence the drug's labeling or promotion. However, data from such studies may subsequently be submitted to FDA for that purpose and, therefore, FDA has an interest in helping to ensure that these studies are designed to yield data adequate to support a labeling change. A Sponsor who would like to obtain FDA advice on study design can submit an IND for FDA review.
Is the Risk Associated With the Product Significantly Increased (or the Acceptability of the Risk Significantly Decreased)?
Investigators should carefully consider the risk implications of any conditions of use in the study that deviate from the conditions of use described in the drug's labeling, with particular attention to the following:
Route of Administration
A change in the route of administration can introduce a significant new risk. For example, there could be a significant increase in risk if a marketed drug for oral administration is converted to a dosage form that is to be administered by injection or intravenous, intrathecal, or inhalation route. These other routes of administration introduce concerns with increased local concentrations, sterility, pyrogenicity, hypersensitivity (e.g., airway reactivity), variations in metabolism, and other issues not present with oral administration that can significantly increase the risk, or decrease the acceptability of the risk, associated with use of the drug.
Dose
Increases in dose, frequency, or duration of administration, compared to labeled dosing regimens, can significantly increase the risk in a study using a marketed drug.
It is also possible that a decrease in dose could significantly increase risk. For example, administering a sub-therapeutic dose of an antiviral drug to study subjects could induce resistance in the subjects, thus rendering a subsequent therapeutic dose of the drug ineffective in treating the virus.
The significance of changes in dose (in particular, increases in dose) can vary across therapeutic areas. For example, the cancer treatment guidance provides some latitude for conducting studies of high-dose cancer treatments without an IND because oncologists are generally familiar with the implications of high-dose regimens. In other clinical settings, use of higher doses than are recommended in labeling may be much more likely to significantly increase the risk or decrease the acceptability of the risk.
Patient Population
The acceptability of known and unknown risks can vary across different treatment populations (see §312.2(b)(1)(iii)).
The population chosen for study could be at increased risk compared to the approved use population for a variety of reasons, such as increased age, different disease or stage of disease, concomitant illness, decreased renal or hepatic function, or concomitant therapy.
For example, a drug with significant toxicity can be approved for use in a population with a life-threatening or severely debilitating disease because the risk of toxicity is acceptable in that population. Use of that drug in a clinical investigation in a population that is not so ill (e.g., to evaluate the drug for prevention of disease or symptomatic relief), however, would present a different risk-benefit situation in which the known risks might not be acceptable. When use of the drug in a specific patient population decreases the acceptability of the known risks, the study would have to be conducted under an IND as required under 21 CFR part 312.
The FDA has issued guidance intended to assist clinical investigators, sponsors, sponsor-investigators, and institutional review boards (IRBs) in determining whether research studies involving human subjects must be conducted under an investigational new drug application (IND), as described in 21 CFR part 312. It is the initial responsibility of the sponsor to determine whether a study meets the exemption criteria listed above. If the sponsor is confident that the study meets IND exemption criteria, the study may be submitted to the IRB. Sponsors who are uncertain if their investigation meets the exemption criteria may seek advice from the FDA by submitting a formal or informal IND exemption request to the FDA.
If the IRB does not agree with the sponsor that the study is eligible for IND exemption, the FDA will need to make the final decision. A formal request for a Type B meeting with the FDA can also help in determining next steps:
- Informal IND Exemption Request: An informal IND exemption request by phone or email can be made to the reviewing division at FDA. However, it is up to the division if they are willing or able to provide feedback informal, or if a full IND submission is needed.
- Formal IND Exemption Request: At minimum, the FDA may require a formal process for requesting an IND exemption by submitting a complete final protocol along with a request and rationale for the exemption in the cover letter. Contact your institute’s regulatory personnel to help with this submission.
Pre-IND Meeting: In some instances, a request for a formal meeting with the FDA, referred to as pre-IND meeting that falls under the category of a Type B meeting, can be requested prior to the submission of the initial IND. Generally, these meetings are not utilized to make exemption requests and are requested to review and reach agreement on the design of animal studies needed to support human clinical testing. However, during this meeting, the FDA may conclude that an exemption is permissible. The format of the IND and the scope and design of planned Phase 1 clinical studies may also be discussed (21CFR312.82).